
Comprehensive characterization of plasmid and AAV gene therapy products with Forge Biologics’ hybrid sequencing approach
Recombinant adeno-associated viral (rAAV) vectors remain at the forefront of the gene therapy revolution, offering hope for treating patients with genetic disorders. Developing assay panels for rAAV drug products to evaluate their potency, purity, and identity reliably is essential to accelerating the availability of these therapeutics to patient communities. Precise sequence identity testing of the plasmid starting materials used in rAAV manufacturing or the genetic material encapsidated within an rAAV vector can be accomplished with a multifaceted sequencing approach, such as that implemented at Forge Biologics.
Historically, the gold standard for DNA sequencing has been chain-termination sequencing (also known as Sanger sequencing). While this method allows for sequencing of moderately long (<1 Kb) DNA fragments with reasonable accuracy, it requires a priori knowledge of targets to sequence heterogeneous populations. Adaptation of sequencing methods for use with gene therapy products requires alternative approaches that allow comprehensive and high-quality characterization of viral vectors and plasmid DNA while maintaining high throughput.
Forge has developed a state-of-the-art hybrid sequencing approach for comprehensive analysis of rAAV vectors and plasmid DNA, overcoming typical platform limitations such as gaps in coverage due to high GC content or low complexity sequences. Interpretation of sequencing data as a quality measure for raw materials and gene therapy products requires specialized analysis. Forge’s in-house analytical team provides a multi-step bioinformatics analysis for each gene therapy product, ensuring that sequencing reads are correctly assigned to their source genomes or sequences with a high degree of confidence. The developed methods allow for efficient sequencing and analysis, cutting down the time to results by 75% compared with traditional outsourced sequencing assays.
Limitations of current sequencing technologies in cell and gene therapy
Advances in sequencing methodology over the past decade have allowed for applications of genomic characterization in cell and gene therapy development, manufacturing, and product release testing. Specifically, sequence identity testing is now often performed as part of characterization testing for both rAAV vectors and high-quality plasmid DNA used as raw materials for gene therapy manufacturing.
When characterizing rAAV vectors and plasmid DNA, both short- and long-read sequencing methods offer unique advantages and limitations. Short-read sequencing, also known as next-generation sequencing (NGS), involves reading short fragments of DNA, typically around 100-600 base pairs in length. This method is known for its high accuracy and high coverage depth, low error rates, and relatively low cost per base. As a result, short-read sequencing is suitable for identifying single nucleotide variants and small insertions or deletions in the rAAV or plasmid genome. The high coverage depth of this sequencing method also makes it helpful in detecting low-frequency variants in AAV or plasmid populations. However, one of the main limitations of short-read sequencing is the difficulty in resolving repetitive regions such as inverted terminal repeats (ITRs) and complex structural variations due to the short-read length.
On the other hand, long-read sequencing, also known as third-generation sequencing, enables the sequencing of a single molecule of DNA, often thousands to tens of thousands of base pairs in length. These modern platforms can overcome limitations found in other sequencing methods, such as short reads, reading errors, or artifacts introduced during fragmentation, amplification, or bench work for sample preparation. Long-read sequencing is beneficial for resolving complex genomic regions, including repetitive sequences or structural variations. It can facilitate the readout of full-length rAAV genomes, including ITRs and flanking sequences [1,2]. However, long-read sequencing methods may have higher error rates compared to short-read sequencing, and the cost per base can be relatively higher.
Gene therapy developers and manufacturers often choose between the two sequencing methods by balancing factors such as accuracy, cost, read length, the complexity of the genomic regions to be sequenced, and based on the material to be sequenced (e.g., plasmid DNA vs. rAAV).
Comprehensive characterization of rAAV vectors through a hybrid short – and long-read sequencing approach
Employing sequencing-based approaches for rAAV vector characterization can provide valuable insights during process development and into clinical and commercial manufacturing. Complete and accurate sequencing can enhance vector design, uncover previously hidden packaging challenges, and provide crucial quality and safety data as part of rAAV release testing.
Forge’s optimized hybrid short- and long-read DNA sequencing approach leverages the strengths of each platform to provide comprehensive and highly accurate sequencing data for gene therapy products (Figure 1A). Utilizing both short- and long-read sequencing provides orthogonal validation of sequencing data, allowing for consistent coverage throughout the genome, even in challenging regions (Figure 1B). While short reads can capture areas of genetic variation, long-read sequencing allows for the detection of complex structural variants, such as inversions, duplications, deletions, or translocation. In addition, short-read sequencing is limited in the ability to fully characterize the rAAV genome, a critical quality attribute being the identity of the gene therapy product. Conversely, long-read sequencing enables quantitative and qualitative characterization of full, partial, and empty rAAV genomes (Figure 1C). In fact, empties always have some genomic content, and long-read sequencing revealed what were previously thought to be empty capsids to contain ITR-bearing short DNA fragments.
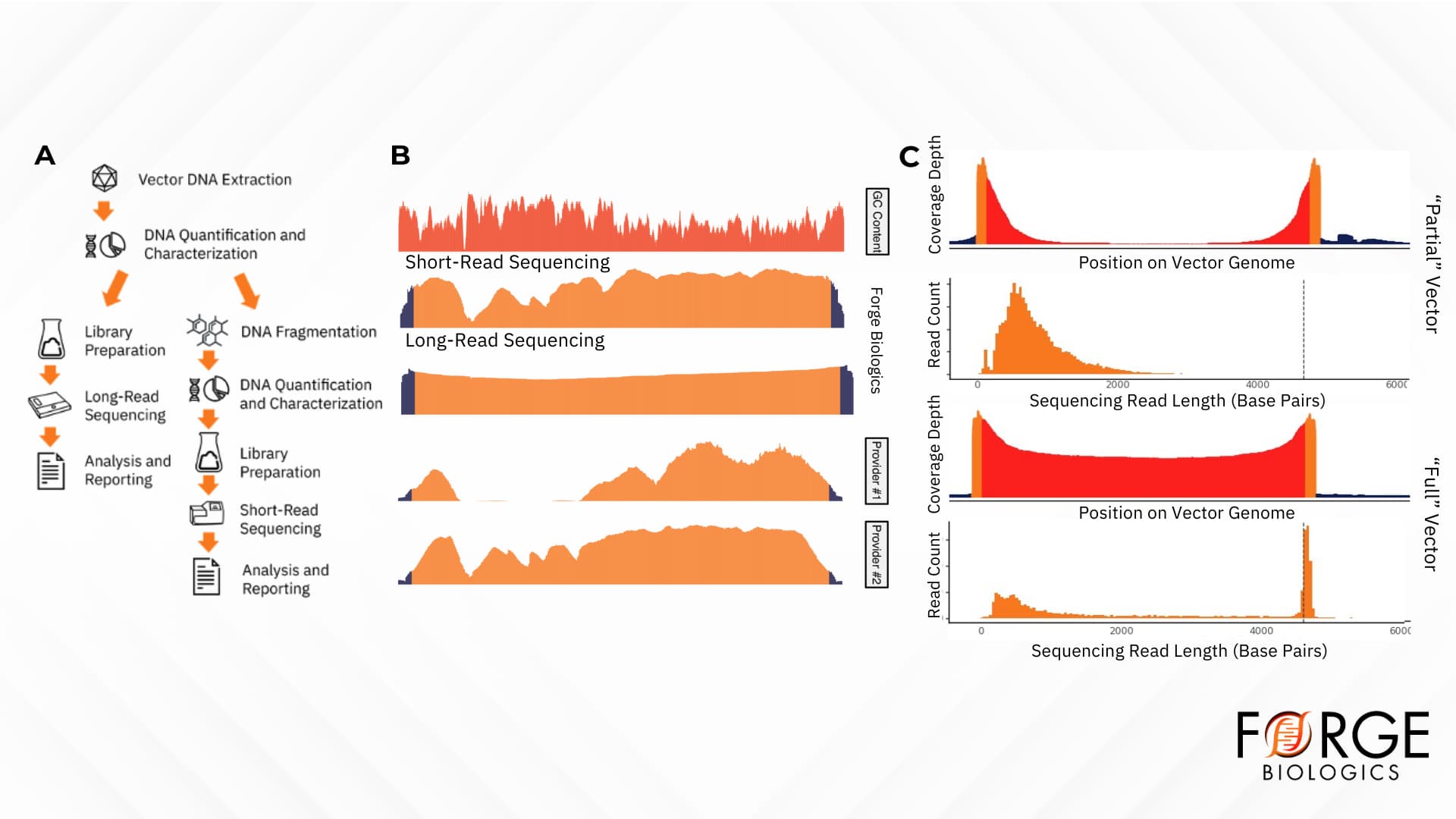
Figure 1. An innovative hybrid short- and long-read sequencing approach for rAAV characterization. A) Graphical schematic of Forge Biologics’ hybrid short- and long-read approach to rAAV sequencing. B) A graphical representation of sequencing coverage of an rAAV product. The top track corresponds to the GC content of the vector, whereas the bottom four tracks represent three short-read sequencing approaches and long-read sequencing of the vector. C) Visualizations of the differences between “partial” and “full” bands of an rAAV product, with the partial band exhibiting a high proportion of short, ITR-adjacent fragments.
In addition to confirming genome sequence identity of rAAV vectors, a hybrid short- and long-read sequencing method can screen for non-target sequences (process-related impurities), such as residual plasmid DNA and residual mammalian host cell DNA (Figure 2A & 2B). Sequence analysis of rAAV vectors can further ensure that oncogenic element sequences (e.g., E1A/E1B, SV-40 large tumor antigen, etc.) are not present in the product (Figure 2B). Forge’s hybrid sequencing approach has high sensitivity, with the ability to detect residual contaminants down to 0.01% in the final product (as demonstrated by spike-in experiments, Figure 2C).
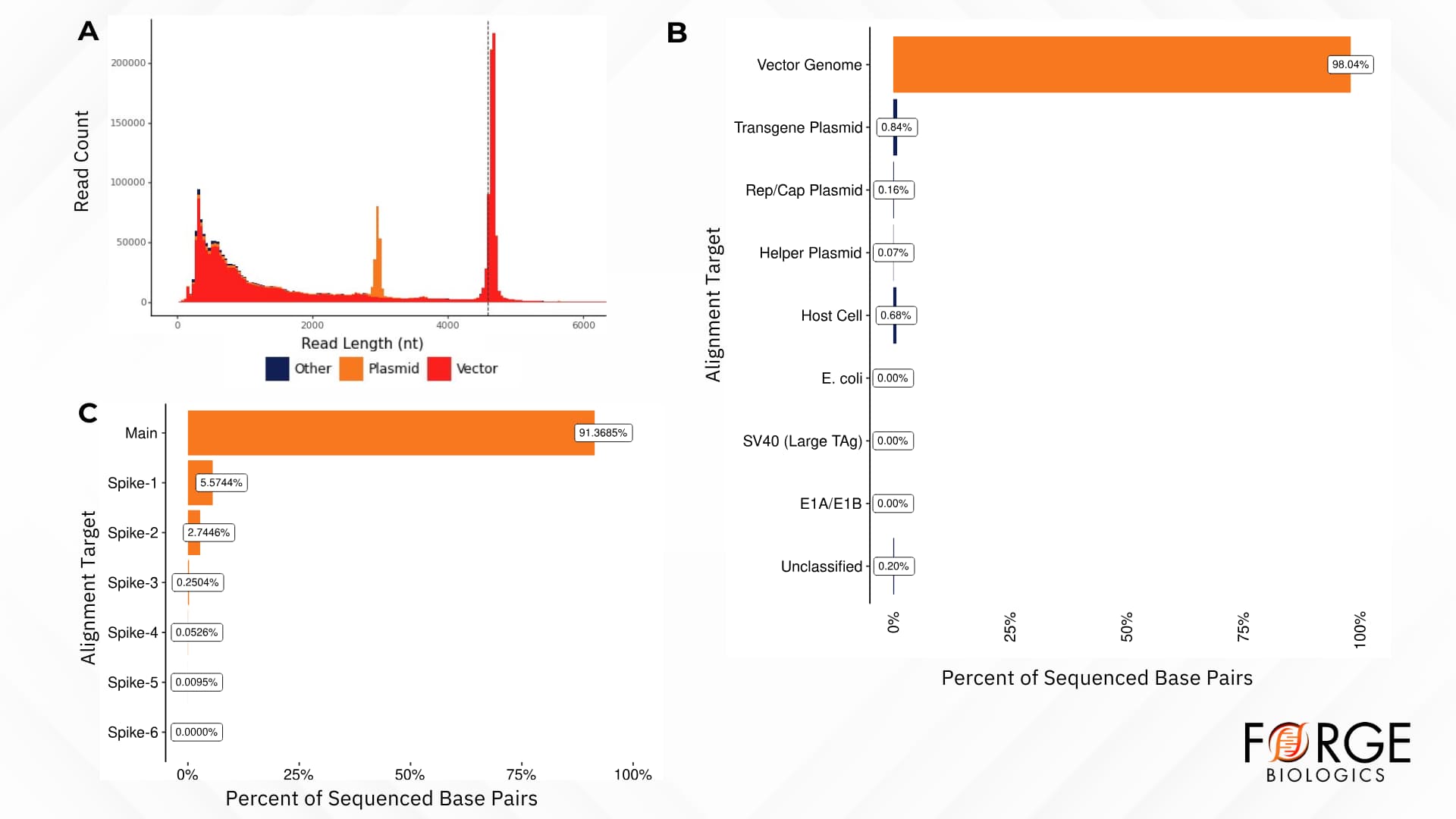
Figure 2. Analysis of impurities in rAAV products using a sequencing-based approach. A) Number and length of reads in rAAV sample aligning with rAAV vector genome (red), residual plasmid (orange), or other impurities (blue). B) A representation of the genome composition of an rAAV product lot, quantifying the percentage of vector genomes and residual impurities in the sample. C) A spike-in experiment using a main plasmid and a set of six “contaminant” vectors, showing detection down to < 0.01%.
In-depth characterization of plasmid DNA by long-read sequencing
Production of high-quality rAAV starts with utilizing high-quality plasmid DNA for triple transfection during the upstream AAV manufacturing process. Ensuring sequence identity is a critical aspect of plasmid DNA characterization and quality control. The high GC content and palindromic nature of ITRs makes gene of interest (GOI) plasmids susceptible to mutations in these regions during plasmid production. Mutations in the ITR region can impact rAAV production [3]. Therefore, it is imperative to confirm the sequence of ITR regions in GOI plasmids prior to use in upstream AAV manufacturing.
Forge Biologics employs a long-read sequencing-based analysis pipeline to rapidly characterize GMP-Pathway and GMP grade plasmids produced using Forge’s plasmid manufacturing services (Figure 3A).
Incorporating sequencing-based analytics into multiple steps of plasmid manufacturing workflows ensures that only high-quality and high-purity materials are used for downstream applications (e.g., rAAV manufacturing). A particular challenge in manufacturing ITR-containing plasmids is the possibility for spontaneous generation of sub-populations of bacteria harboring truncated ITRs (Figure 3B). From the initial generation of bacterial master cell banks, long-read sequencing checkpoints ensure even coverage, and monitor ITR integrity and stability throughout culture and plasmid production, with the ability to detect the presence of any sub-populations containing truncated ITRs (Figure 3C-D).
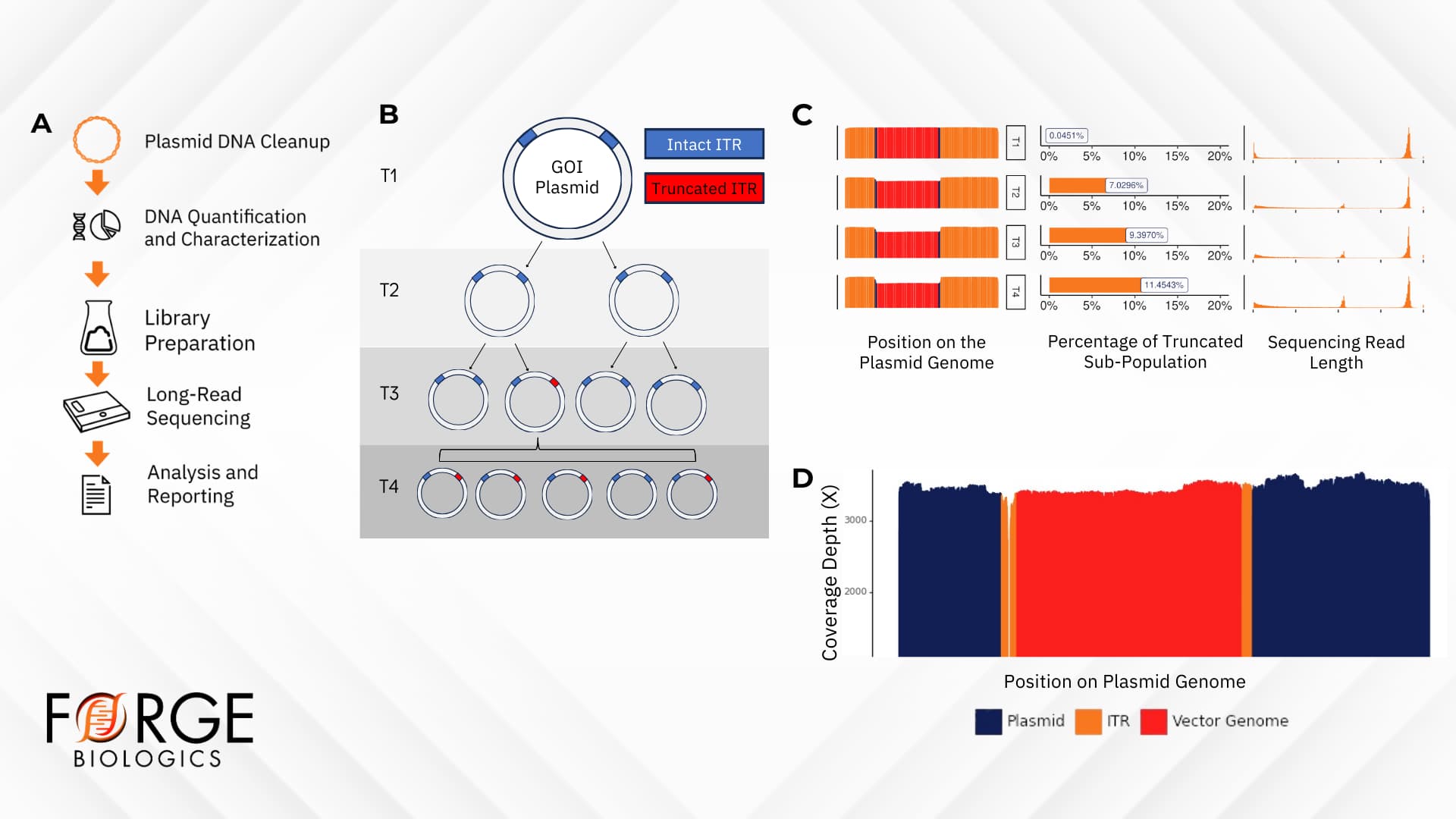
Figure 3. Long-read sequencing of high-quality DNA ensures the absence of sub-populations in bacterial cell banks. A) Schematic of plasmid DNA preparation and sequencing workflow. B) Graphical representation of spontaneous generation of bacterial sub-populations containing truncated ITRs. The GOI plasmid contains intact ITR regions (blue) at the initial timepoint (T1) post-transformation into a bacterial host (e.g., E. coli). As the bacteria is passaged (T2-T3), sub-populations may acquire spontaneous mutations resulting in a truncated ITR region (red) in the GOI plasmid. The proportion of the bacterial population harboring a truncated ITR may increase across passages (T4). C) Visual representation of a plasmid across four timepoints. A truncated subpopulation can be observed growing as timepoints advance, with the graphical representations showing the coverage across the plasmid genome, the observed truncation percent, and a histogram of read lengths showing a secondary peak appearing, corresponding to the truncated (shorter) subpopulation. D) Graphical representation of coverage across a plasmid genome, showing even coverage across most of the genome, with a noted deletion in the 5’ ITR sequence.
Forge Biologics’ state of the art hybrid sequencing approach for plasmid and rAAV gene therapy products
Currently available sequencing solutions have required significant development and optimization to be compatible with gene therapy products. However, proper sequencing of plasmid starting materials and rAAV products is critical for maintaining product quality and safety. Forge’s hybrid short- and long-read sequencing approach allows for adaptable, reliable, and comprehensive characterization of products, including the detection of residual impurities with high sensitivity (Figure 4). The orthogonal validation that Forge’s hybrid short-and long-read genome sequencing workflow provides inherently increases confidence in the safety and quality of gene therapy products.
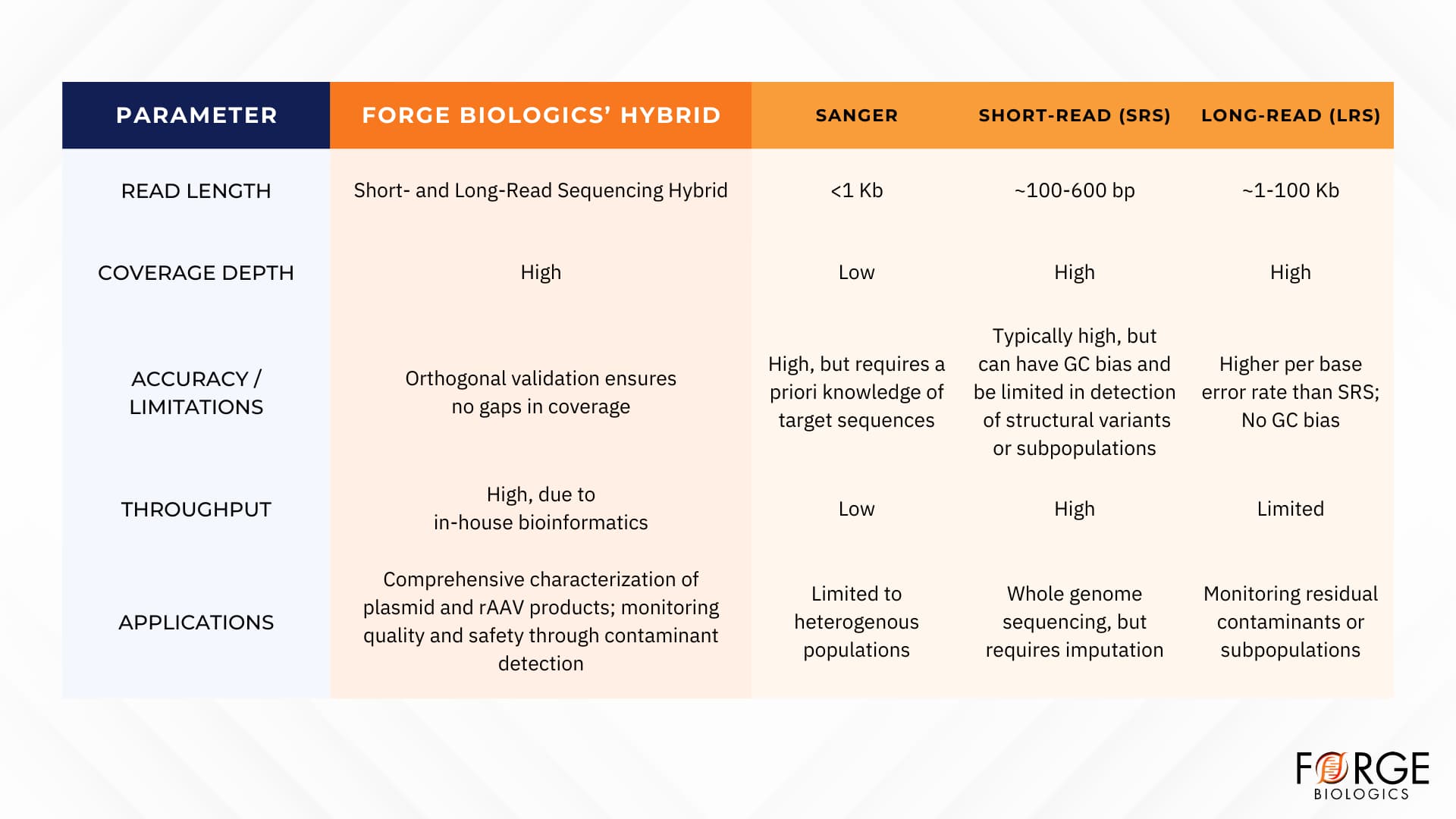
Figure 4. Overview of sequencing-based methods for plasmid and rAAV gene therapy products
Learn more about Forge Biologics’ state of the art hybrid sequencing approach for plasmid and rAAV gene therapy products and connect with our team here.
References:
1. Tai PWL, Xie J, Fong K, Seetin M, Heiner C, Su Q, Weiand M, Wilmot D, Zapp ML, Gao G. Adeno-associated Virus Genome Population Sequencing Achieves Full Vector Genome Resolution and Reveals Human-Vector Chimeras. Mol Ther Methods Clin Dev. 2018 Feb 13;9:130-141. doi: 10.1016/j.omtm.2018.02.002. PMID: 29766023; PMCID: PMC5948225.
2. Namkung S, Tran NT, Manokaran S, He R, Su Q, Xie J, Gao G, Tai PWL. Direct ITR-to-ITR Nanopore Sequencing of AAV Vector Genomes. Hum Gene Ther. 2022 Nov;33(21-22):1187-1196. doi: 10.1089/hum.2022.143. PMID: 36178359; PMCID: PMC9700346.
3. Assad W, Volos P, Maksimov D, Khavina E, Deviatkin A, Mityaeva O, Volchkov P. AAV genome modification for efficient AAV production. Heliyon. 2023 Apr 1;9(4):e15071. doi: 10.1016/j.heliyon.2023.e15071. PMID: 37095911; PMCID: PMC10121408.
Contributing Author: Esko Kautto, Ph.D., Senior Scientist, Analytical Development, Forge Biologics

