Five strategies for mitigating the impact of COVID-19 on clinical trials
By Jenny Gidley, Vice President, Global Clinical Operations, Alberto Grignolo, Corporate Vice President, Bridget Heelan, Vice President, Regulatory and Access Consulting Group, and Amy McKee, Vice President, Regulatory Consulting Services — Parexel International
COVID-19 has brought with it a number of challenges in managing ongoing clinical trials for chronic, life-threatening, and rare diseases. Enrollment is slowing or even stopping in some cases to prioritize patient safety; travel restrictions are preventing in-person patient site visits and CRA monitoring visits; regulators and industry staff who once traveled widely are working from home; some IRBs are deferring protocol review meetings, and investigator sites and hospitals are justifiably prioritizing care over clinical research activities.
However, to ensure that patients – especially those where clinical research is really their only care option
– are supported as much as possible, companies can navigate this crisis successfully by re-examining and adapting their clinical research operations to ensure patient safety, increase communications with regulators, assess risks, preserve data integrity, and comply with fast-changing regulations.
Regulators are responding with flexibility and pragmatism
Regulators worldwide have mobilized to streamline requirements for developing diagnostics and products to treat COVID-19. For example, the FDA’s new Coronavirus Treatment Acceleration Program (CTAP) (launched March 31) is reviewing COVID-19 study protocols within 24 hours, approving single- patient expanded access requests in three hours, and analyzing the use of real-world data (RWD) for illness patterns and treatment outcomes.
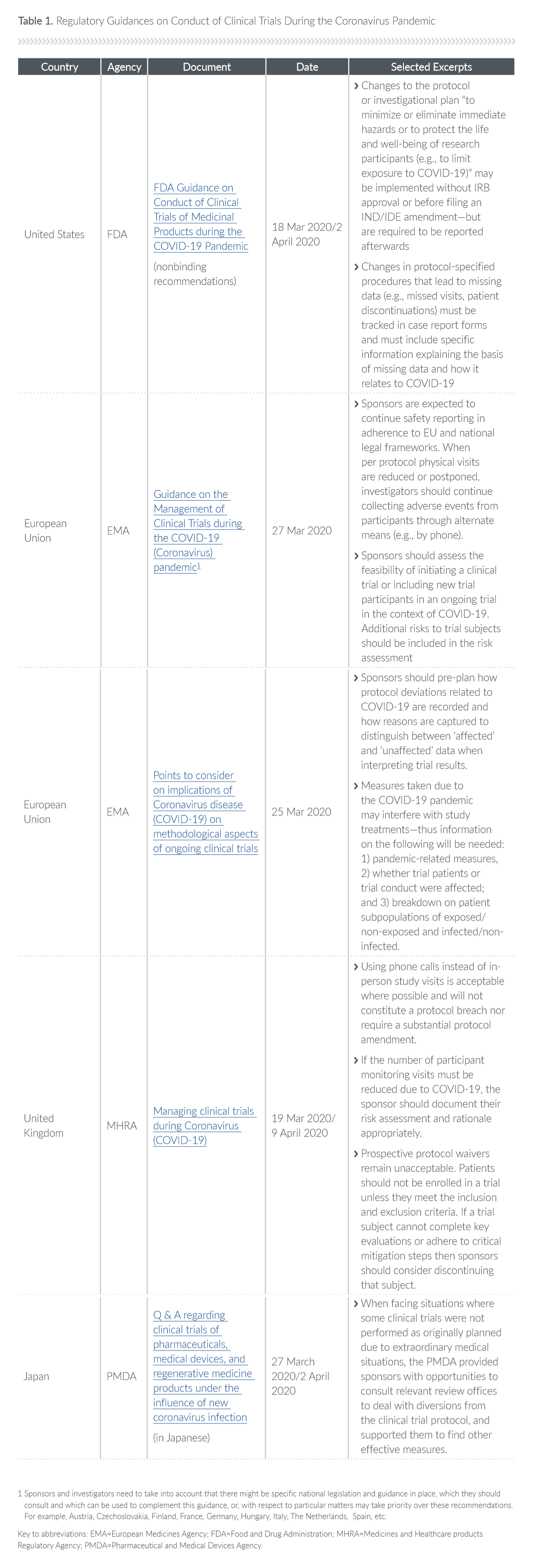
But what about non-COVID-19 trials? Agencies have issued guidance documents for companies on how to conduct trials during the coronavirus pandemic (see Table 1), but we are in uncharted territory.
Sponsors face complex choices, among them extending the duration of trials, halting or postponing trials or closing trial sites, transferring patients from sites hit hard by COVID-19, conducting procedures at new locations, suspending the enrollment of new patients, and modifying trial protocols.
Disruptions could lead to a new normal
New technologies, such as risk-based monitoring, eConsent, wearables, telemedicine patient visits, and remote source document verification, are being deployed and pressure-tested in a clinical research environment unimaginable just a few months ago.
Although study timelines will change, enrollment may quickly rebound once sites reopen. For example, Parexel is already seeing some recovery in Asia-Pacific countries like China, where activities are resuming, as are our on-site monitoring visits. How timelines will ultimately be affected will depend on how long stay-at-home orders persist in many different places.
New best practices adopted now to adapt to and mitigate the disruptions of the current crisis will likely have a shelf life beyond it. The speed, efficiency and cost savings of virtual trials (that leverage digital health technologies and allow patients to participate from home), and hybrid trials (that combine randomization and blinding with real- world data collection), could alter the clinical trial landscape permanently.
To that end, sponsors, CROs, investigators, sites, regulators, and patients should focus on developing and refining practices that will allow clinical trials to continue, and that will improve the efficiency and outcomes of clinical research in the future.
Five strategies to help navigate the current crisis while improving future clinical trials:
1. Ensuring patient safety with remote monitoring
The priority of drug development has always been patient safety. As a result, the industry has quickly pivoted towards remote solutions for the safety challenges to clinical trials posed by COVID-19 social distancing and stay-at-home orders.
In the current environment, sponsors will not be able to monitor studies in the traditional way. In most instances, clinical research associates (CRAs) or trial monitors will not be able to travel to sites to review, verify, and discuss study data. The informed consent process, adverse event reporting and review, investigational product handling, and data monitoring – tasks that are normally done on-site in a conventional trial – will now have to be accomplished remotely, via telephone or electronic communication, in a structured manner. This represents a sea change, and a significant challenge, for sponsors and regulators.
At Parexel, we have been able to implement procedures for remote monitoring visits by leveraging our experience in late-phase studies and applying it to earlier Phase II and III studies. We are exploring new methods to access source documents remotely, and investigators are finding ways to interact with patients via phone or video (often referred to as telehealth or telemedicine). We are sending clinical trial materials – including the study drug when possible – directly to patients if those patients are not able to come to the site.
Of course, if an investigator or sponsor cannot run a study in compliance with the protocol, or monitor patient safety virtually with complete confidence, they will need to pause a study, halt enrollment, or discontinue a participant. For example, on March 22, due to safety concerns regarding immunomodulatory agents, Galapagos NV paused enrollment in the Phase II and III trials of its JAK1 inhibitor filgotinib in Crohn’s disease, psoriatic arthritis, inflammatory bowel disease, uveitis, and other inflammatory conditions. Pausing enrollment allows sites to focus on the safety of those patients already enrolled in studies, and maintain continuity of treatment.
The safety of patients can be protected remotely. However, solutions will need to be customized to a trial’s patient population, protocol, and geography, among other factors.
2. Taking a new approach to risk assessment in a time of new risks
Everyone is struggling with the risk assessment aspect of modifying the protocols of ongoing clinical trials. How will protocol changes impact patient safety, data integrity, and interpretation of results?
Risk assessment is a highly granular process. Each issue must be assessed for each patient in each trial in each country. That demands attention to detail and deep expertise. It also adds expense. There are few generic answers to the new risk assessment problems that have arisen in the time of COVID-19, but we have found there are workarounds that are effective in a majority of clinical trials, even some with complex designs.
Sponsors must assess both the risk of COVID-19 potentially affecting study participants directly (for example, if they fall ill with the disease), and whether COVID-19 related measures (such as social distancing) will affect trial conduct.
The recent EU guidance on COVID-19 (see Table 1) requires the sponsor of each trial to perform a COVID-19-driven benefit-risk evaluation with risk mitigation measures, ideally as an amendment to the benefit-risk section in the protocol. This benefit-risk assessment should include:
- Accounting for the additional risks posed to participants by the COVID-19 pandemic, along with risk mitigation measures taken;
- How the risks of involvement in the trial (factoring in COVID-19) are weighed against anticipated benefits for the participant and society. In case these two conflict, subject safety always prevails;
- The relevant parties’ input (for example, the medical monitor’s), documented on an ongoing basis, prioritizing critical tasks in the clinical trial and how those are best executed.
Building out the expertise and finding the bandwidth to conduct comprehensive risk assessments for ongoing clinical trials should be an even greater priority for sponsors today.
3. Communicating in new ways with regulators who are listening
Once risk mitigation strategies and risk assessments of clinical trial changes are underway, sponsors must inform regulators through any means possible, even by email or telephone, and be open with them about potential problems they have identified.
Today, developing COVID-19 vaccines and treatments is a global priority. The FDA is prioritizing submissions for COVID-19-related products. To achieve that, they are redeploying staff from less busy areas, or those who have prior experience regulating a product that has been repurposed for treatment of COVID-19. Although the redeployment has not created delays in review activities to date, delays remain a real possibility.
In the EU, face-to-face meetings and scientific advice in person are no longer common. It is thus increasingly important to interact with and solicit input from regulators by phone and email. But policies on new ways to collect data vary by country. For example, some member states are allowing centralized monitoring of clinical trials and source data verification as long as patient confidentiality is maintained. However, the electronic transfer of data is not broadly accepted: it is possible in some European countries and not in others. The key is discussing it with regulators prior to doing it.
The FDA is showing remarkable flexibility on a range of issues, from how a sponsor may document informed consent, to conducting clinical trials virtually (see Table 1), to using real-world evidence in a marketing submission. This gives the industry permission to test the boundaries. If an unprecedented solution can meet regulatory requirements, as well as the needs of patients and sponsors, regulators may be more open to it today than in the past.
Although regulators will never compromise on patient safety, or on the need to show efficacy with GCP-compliant data, there is now flexibility on how and when data are collected (for example, endpoints can be delayed), how study monitoring can be done, and how investigational drugs may be delivered to patients, to name just a few. Regulators need to be informed about any solutions sponsors may want to try, and companies can protect their drug development programs by communicating them effectively.
4. Preserving data integrity under extraordinary conditions
Where sponsors have to modify study protocols in response to the Coronavirus pandemic, regulatory agencies expect meticulous records of deviations in order to preserve the integrity and interpretability of trial data.
Regulators around the world have indicated they will be more flexible than in the past. For example, new FDA guidance (Table 1) specifies that changes to a protocol or investigational plan meant “to minimize or eliminate immediate hazards or to protect the life and well-being of research participants (e.g., to limit exposure to COVID-19)” may be implemented without IRB approval or before filing an IND/IDE amendment—but are required
to be reported afterwards.”
Acknowledging that the disruptions caused by the pandemic may result in an increase in missing data, the FDA is requiring that missed visits, patient discontinuations, and similar changes in protocol- specified procedures must be tracked in case report forms and must include specific information explaining how the missing data relate to the COVID-19 pandemic (see Table 1).
The EMA has encouraged sponsors to “pre-plan” how protocol deviations related to COVID-19 are recorded and how reasons for missing data are captured, in order to distinguish between “affected” and “unaffected” data when interpreting trial results. While the EMA have released an overall guidance for the EU, the National Regulatory Authorities of EU member states are directly responsible for the trials. Accordingly, sponsors need to check country- specific guidance documents (see Table 1).
5. Staying alert to changing regulations; staying engaged
Sponsors will need to track the ever-changing COVID-19 guidelines issued by health authorities and regulators in every locality where they are conducting clinical trials. Regions vary in terms of the stringency of their travel restrictions, their requirements for social distancing, lockdown and quarantine rules, and those for testing and contact tracing.
A large number of clinical trials (though not all) will inevitably be paused until the coronavirus pandemic subsides, but sponsors should stay engaged. For example, Parexel is currently involved in a high volume of regulatory work for clients who are getting everything in line for when their paused trials resume. If the preparatory work is done now, studies can restart more efficiently. This crisis will not last forever.
Tracking regulatory guidances and assessing how flexible different agencies may be will be key to surviving in this temporarily disrupted drug development environment.
Change is a matter of survival
This public health emergency will change clinical trials forever but many of those changes will be for the better. Both industry and regulators are being pushed to explore new ways to conduct clinical trials, discovering the benefits and limits of a more virtual, decentralized approach. This experiment forced upon us will teach us crucial lessons about what works and what doesn’t.
If new pathways and procedures are proven effective during this pandemic, regulatory authorities will likely continue to accept them in the future. After all, the expedited approval pathways and regulations that so many companies use today were written in response to the HIV crisis, when patients needed access to new therapies quickly. Change was a matter of survival then, and it is a matter of survival now.
The legacy of our current public health crisis could be that virtual trials and hybrid trials will become much more widely accepted and that these and other patient-centric study designs will be working to benefit patients and sponsors when the novel Coronavirus has become a matter for historians, and ceased to be one for researchers.
FDA Guidance on Conduct of Clinical Trials of Medicinal Products during the COVID-19 Pandemic (nonbinding recommendations)
18 March 2020 / 2 April 2020
United States, FDA
- Changes to the protocol or investigational plan “to minimize or eliminate immediate hazards or to protect the life and well-being of research participants (e.g., to limit exposure to COVID-19)” may be implemented without IRB approval or before filing an IND/IDE amendment—but are required to be reported afterwards
- Changes in protocol-specified procedures that lead to missing data (e.g., missed visits, patient discontinuations) must be tracked in case report forms and must include specific information explaining the basis of missing data and how it relates to COVID-19