Turning the corner on treating the root cause of sickle cell disease
Early in my career, as a medical resident, I saw first-hand the enormous challenges faced by children and adults with sickle cell disease (SCD), a genetic blood disorder that historically has lacked adequate treatment options. People living with this life-long disease are mainly those with ancestors from sub-Saharan Africa, as well as people of Hispanic, South Asian, Southern European and Middle Eastern descent. These patients suffer from devastating physical symptoms, including progressive, eventually fatal, organ damage and excruciating pain. In addition, they encounter emotional, mental and social burdens – non-physical aspects of living with SCD that also take a serious toll on patients and their caregivers.
As a young clinician, I was often disappointed, angry and even ashamed at how these patients were treated. Indeed, for a long time, SCD has been a severely neglected disease, but recently we have turned a corner in the form of important innovations that are changing how people with SCD are cared for, giving the SCD community hope for the future.
In the more than 100 years since physicians began recognizing SCD as a distinct disease, the care of those affected progressed at a snail’s pace. For most of this time, physicians were limited to managing pain and trying to make their patients as comfortable as possible. In the 1950s and 1960s, a number of discoveries deepened our understanding of this inherited disorder. Most notably, scientists discovered that the root cause of SCD is the polymerization of hemoglobin, the protein in red blood cells that transports oxygen to cells and organs. This molecular process causes red cells to become deformed and take on the sickle shape that is characteristic of the disease. Polymerization ultimately results in hemolysis (the destruction of red blood cells) and anemia, which in turn sets off a cascade of events that can lead to long-term and devastating consequences, including multi-organ failure, stroke and early death.
SCD results in morbidity and mortality via distinct pathways
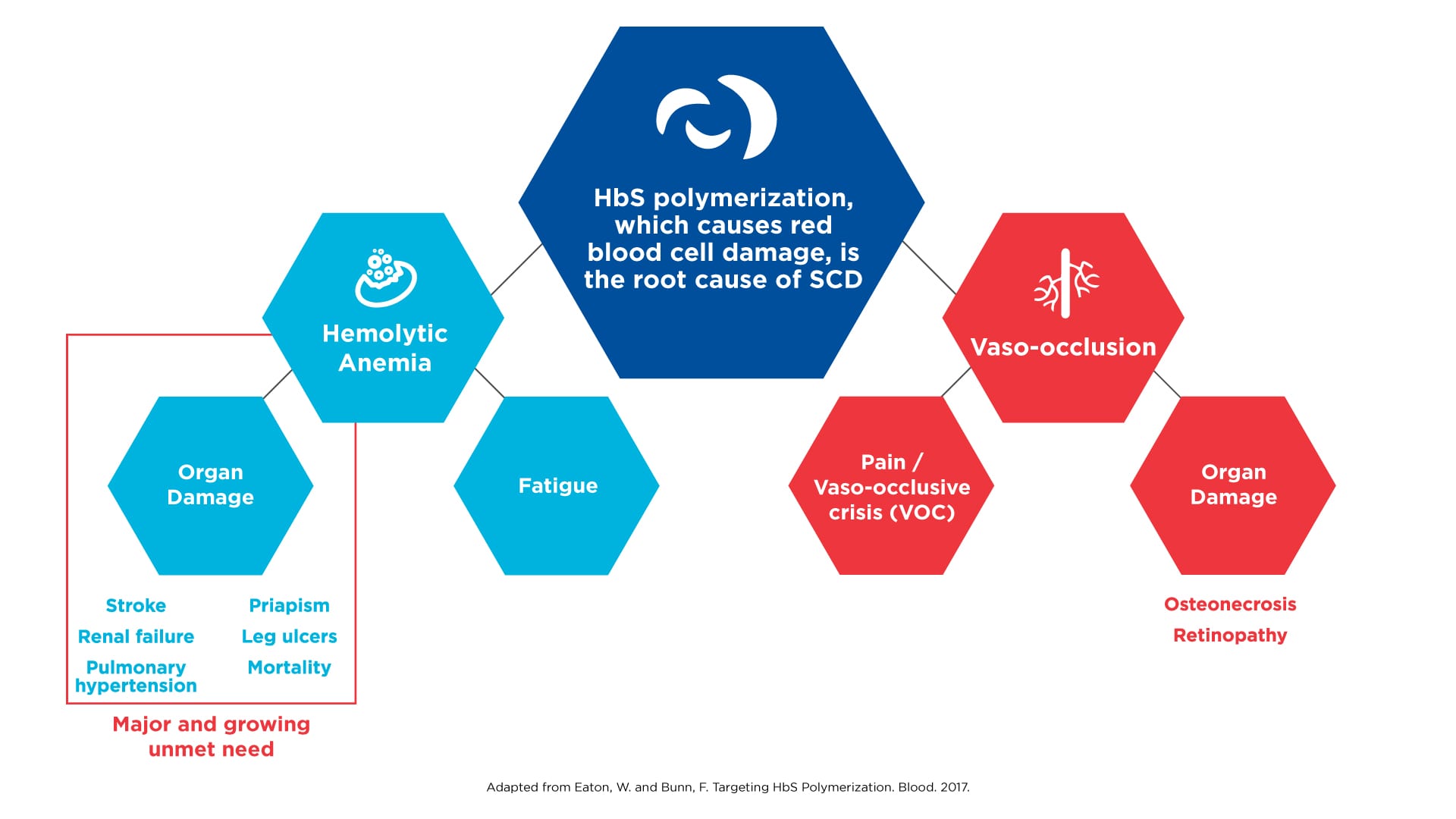
Click on the image to see the full-sized version
Despite this fundamental insight about the molecular driver of SCD, there was a glaring lack of innovation and initiative by the biopharmaceutical industry and academia to apply this discovery to the development of new treatments, as illustrated by the very few medicines for SCD approved by the FDA over the past few decades. Much of the focus continued to be on treating the symptoms and complications of the disease – similar to what we saw in the early days of HIV/AIDS. However, in the last decade, we began to see a renewed interest in SCD. A shift occurred toward developing treatments that are truly disease-modifying. The FDA has kept pace with this trend by changing how it evaluates clinical trial results and approves innovative medicines, so companies can get them to patients in need faster.
Drug development in SCD has lagged other orphan diseases, such as cystic fibrosis and hemophilia
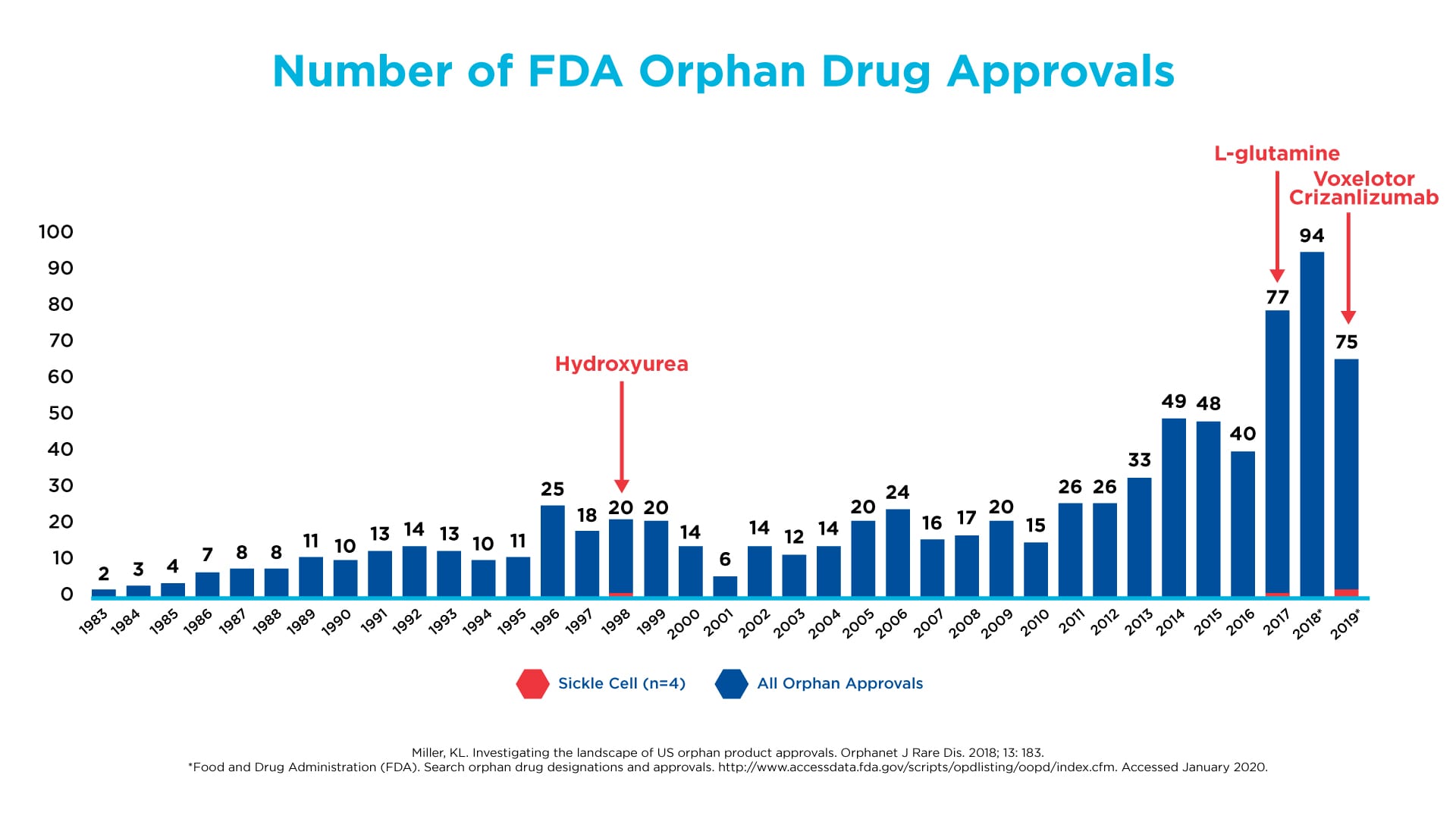
Click on the image to see the full-sized version
At Global Blood Therapeutics (GBT), our focus from the start has been on sickle hemoglobin polymerization, the root cause of the disease. Low hemoglobin is a key marker and risk predictor that has been established in the literature as playing a critical role in clinical outcomes for individuals with SCDᶦ, much like how viral load is a key marker used to evaluate how well people with HIV are responding to anti-viral treatment. Concentrating on this oxygen-carrying molecule, GBT researchers looked beyond symptoms to the underlying biology of SCD. We specifically engineered a molecule to address the scientific challenges posed by SCD.
Today, people with SCD are finally benefiting from the innovation of disease-modifying treatments. Recently, the FDA approved our medicine, Oxbryta™ (voxelotor) tablets, for the treatment of SCD in adults and children 12 years of age and older. Oxbryta is the first FDA-approved medicine that specifically targets the root cause of SCD by directly inhibiting the polymerization of hemoglobin. It works by selectively entering red blood cells and binding to hemoglobin, resulting in an increase in the affinity of hemoglobin for oxygen. This stabilizes the red blood cells in an oxygenated state, preventing polymerization and the resultant sickling and destruction of red blood cells. The regulatory approval of Oxbryta would not have been possible without the steadfast dedication and commitment to collaboration from the medical, scientific and SCD communities, as well as the FDA.
Oxbryta marks a new phase in the evolution of care for people with SCD. While pain is often assumed to be the most common symptom of SCD, the manifestations of this disease vary greatly from person to person. In fact, about half of all people with SCD do not experience pain crises at all.ᶦᶦ The universal commonality among individuals with SCD is that they suffer from hemoglobin polymerization. By stopping this molecular process where it starts, we have the potential to meaningfully modify the course of this serious disease rather than just address the symptoms.
Organ damage in SCD patients

Click on the image to see the full-sized version
The recent transformation in how we approach SCD treatment – with two recent FDA approvals of SCD medicines and ongoing research programs by GBT and other companies – represents a true paradigm shift and makes me more optimistic than ever about the future for the SCD community.
Full Prescribing Information for Oxbryta is available here.
ᶦ Ataga KI, et al. Blood. 2018;132:12.
ᶦᶦ Shah N., et al. Sickle Cell Disease Complications: Prevalence and Resource Utilization. Plos One. 2019. Jul 5;14(7):e0214355.

